In early 2019, we volunteered as part of a group of researchers attempting to create early procedures for COVID-19 field testing. As part of this, we developed several techniques for sequence motif characterization and mining.
One of the tools scans a set of known protein variants in search for space-disparate motifs, corrected for amino-acid similiarity. We look for motifs that characterize a positive set, without matching a negative control set. We also calculate the probability that such motifs will appear, by chance, in the provided sample; then look for low-probability characterizing motifs. Here's an example visualization of motifs in a CRISPR-Cas protein family:
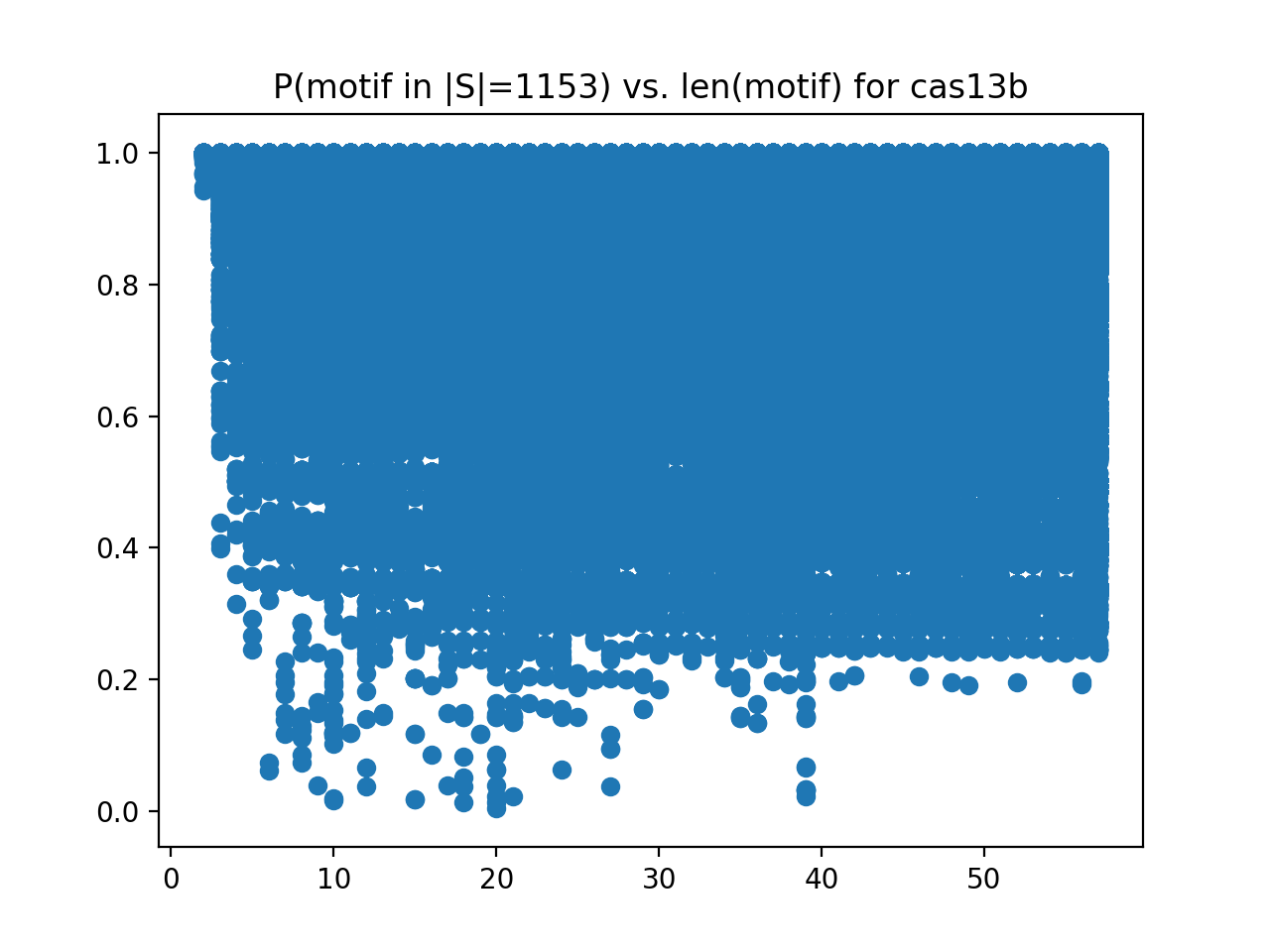
This scatterplot has the length of the motif on the x-axis, and the probability that it occurs in a random family of the same size as the y-axis. The dots indicating length low-probabilty length 39 motifs indicate high-quality motifs for this family, worth further investigation.
We identified several such candidate motifs, and then visualized how they align on particular proteins in the family:
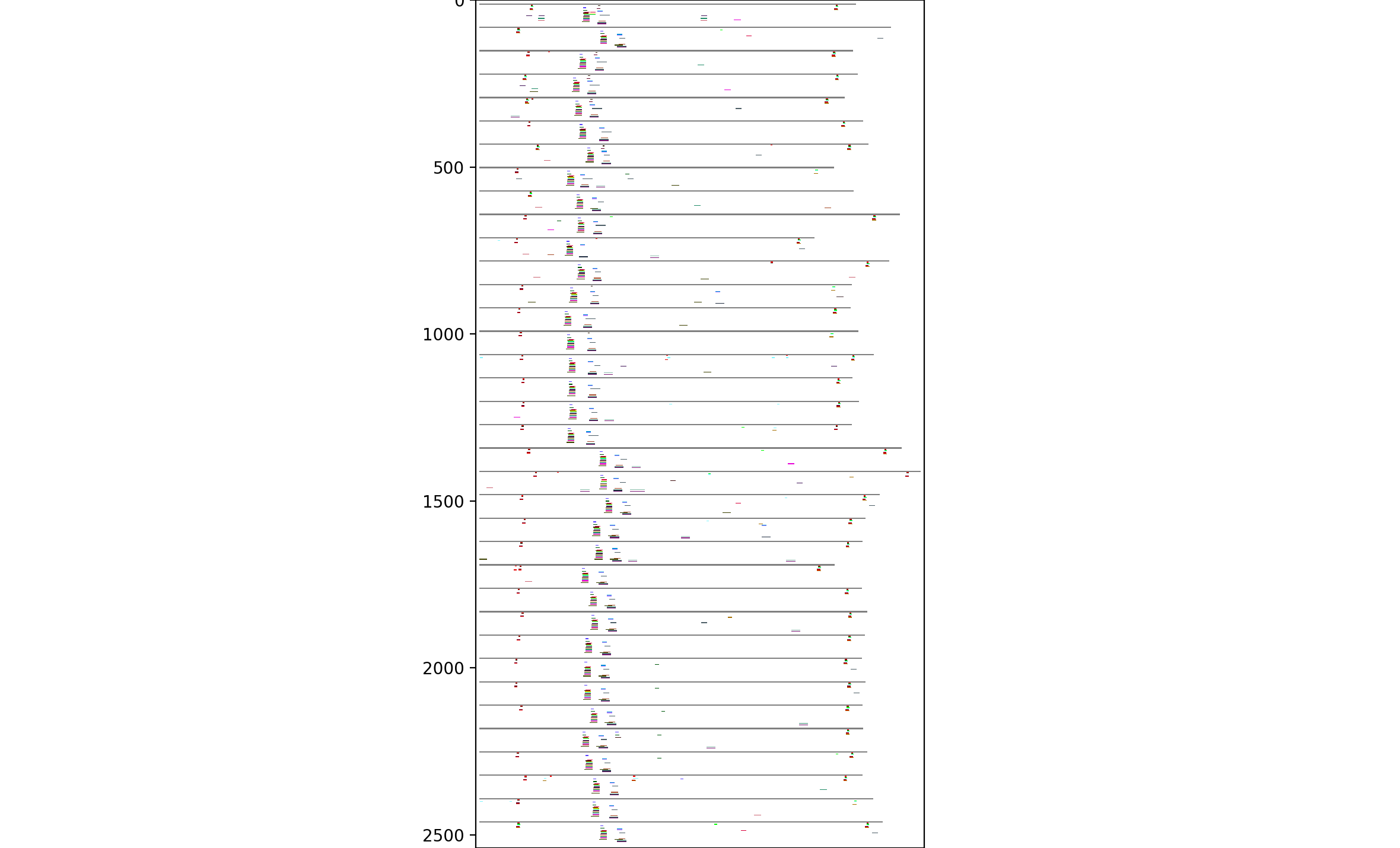
From this, there seem to be four common clusters of motifs: two short ones on the right and left edges, and a pair of consistently-space but shifted motifs in between. Looking at the top two, in more detail:
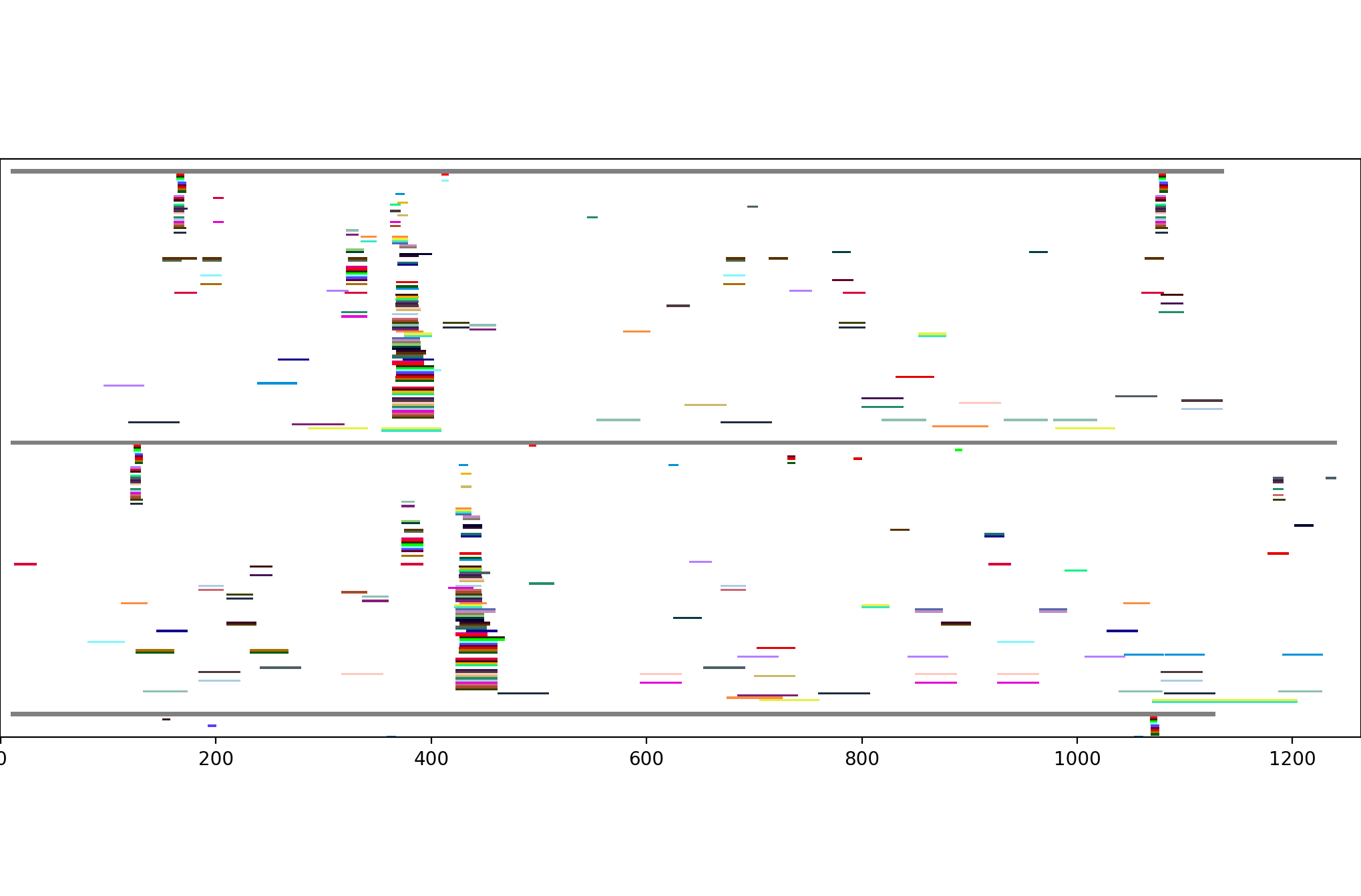
The single-line motifs showing up by themselves indicate random-chance "motifs" -- in other words, in a random sample of this size, one would expect a certain number of these motifs to happen just by chance. But the large, consistent groupings indicate more reliable sequence motifs, which probably correspond to structural motifs in the functioning protein.
Once a combination of sequence motifs has been identified, we can perform lightning-fast searches of popular databases (e.g., BLAST-nr or UniProt) to find other proteins that match the provided motif-query. Such sequences can then be experimentally tested for desired behaviors, and this process iterated to refine the search.
In addition to sequence motifs, we've also developed a number of other sequence characterization tools, including HMM models, clustering metrics, and wavelet models. Different tools emphasize different kinds of similarity, and our in-depth knowledge of the underlying computational and statistical techniques allow us to tune the models best for a specific application.
We also now have libraries that automatically search or download sequences and metadata from a number of public database, including PDB, GeneBank, NCBI, PFAM, InterPro, and Uniprot.