The SPATS package implements a read mapping and reactivity analysis pipeline for calculating SHAPE-Seq reactivities from an input set of next-generation reads. The 496code team has been maintaining this package since 2017.
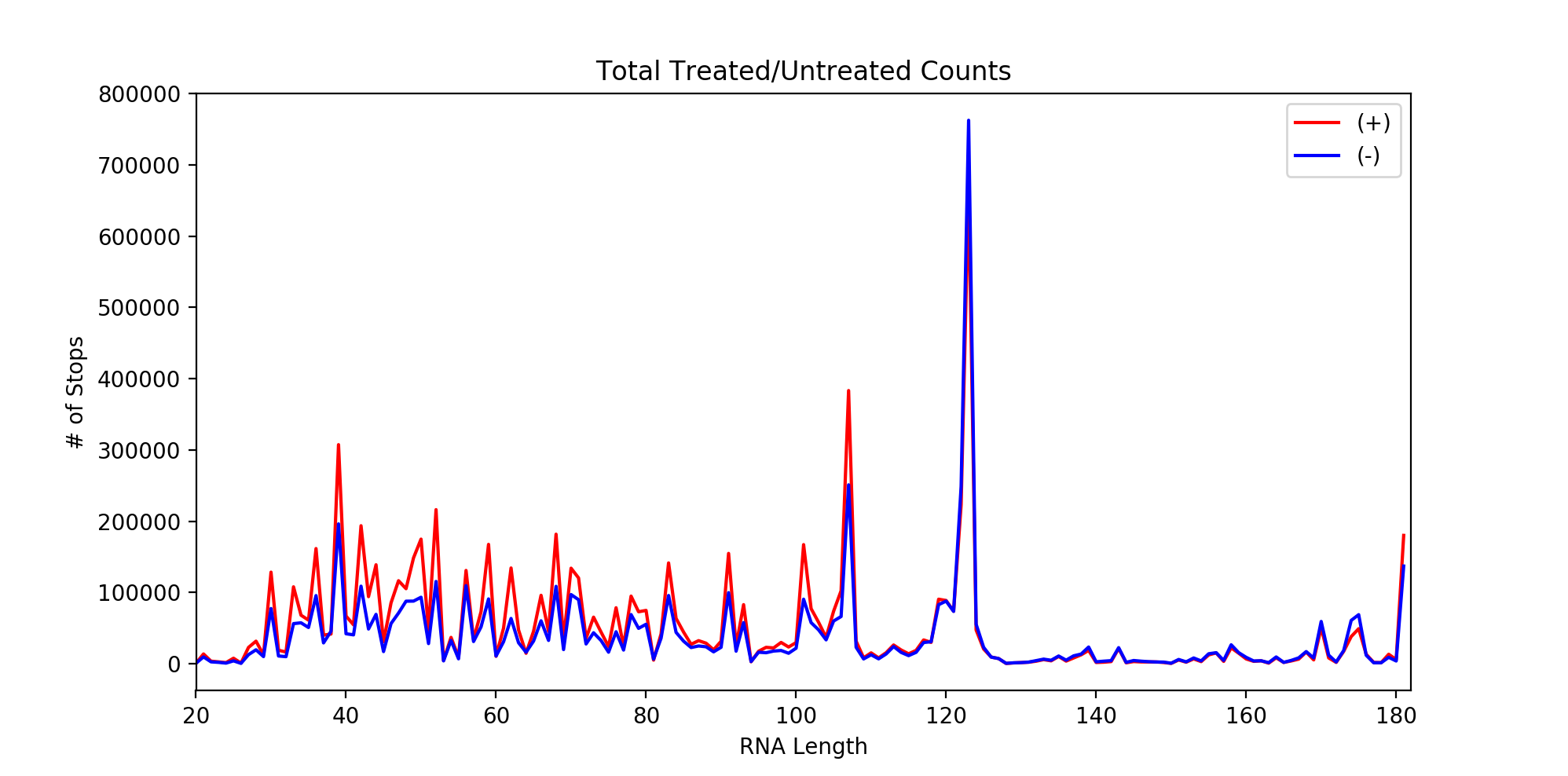
To the right is an example reactivity plot for a co-transcriptional experiment, automatically generated by the software.
An overview of what we've done:
- First task: improve ability for other labs to use SPATS. The original SPATS installation instructions were 400 lines long, not including the required installation of several dependent libraries. The process was known to fail on Mac OSX and most Linux systems (no Windows support).
- After our rewrite of the code, the install instructions consist of one command, and works on any OS. It also includes complete and extensive documentation, with a Getting Started tutorial and suite of command-line tools to automate the most common tasks.
- In addition to improving usability, the new code is also more accurate -- in the process of development and verification, we found and fixed a number of bugs discovered with the original implementation. The current code has a comprehensive set of unit tests, as well as an extensive regression test suite with over 200 specific sequence alignments to ensure that changes to code don't cause known sequence alignments to fail.
- The new code is also several orders of magnitude faster: a standard co-transcriptional analysis that took roughly 3.5 hours on a standard laptop now finishes in under 14 seconds.
- The code has evolved over time to handle updates to experimental procedure. The current code has over 40 configuration options that cover various changes in experiment and analysis options.
-
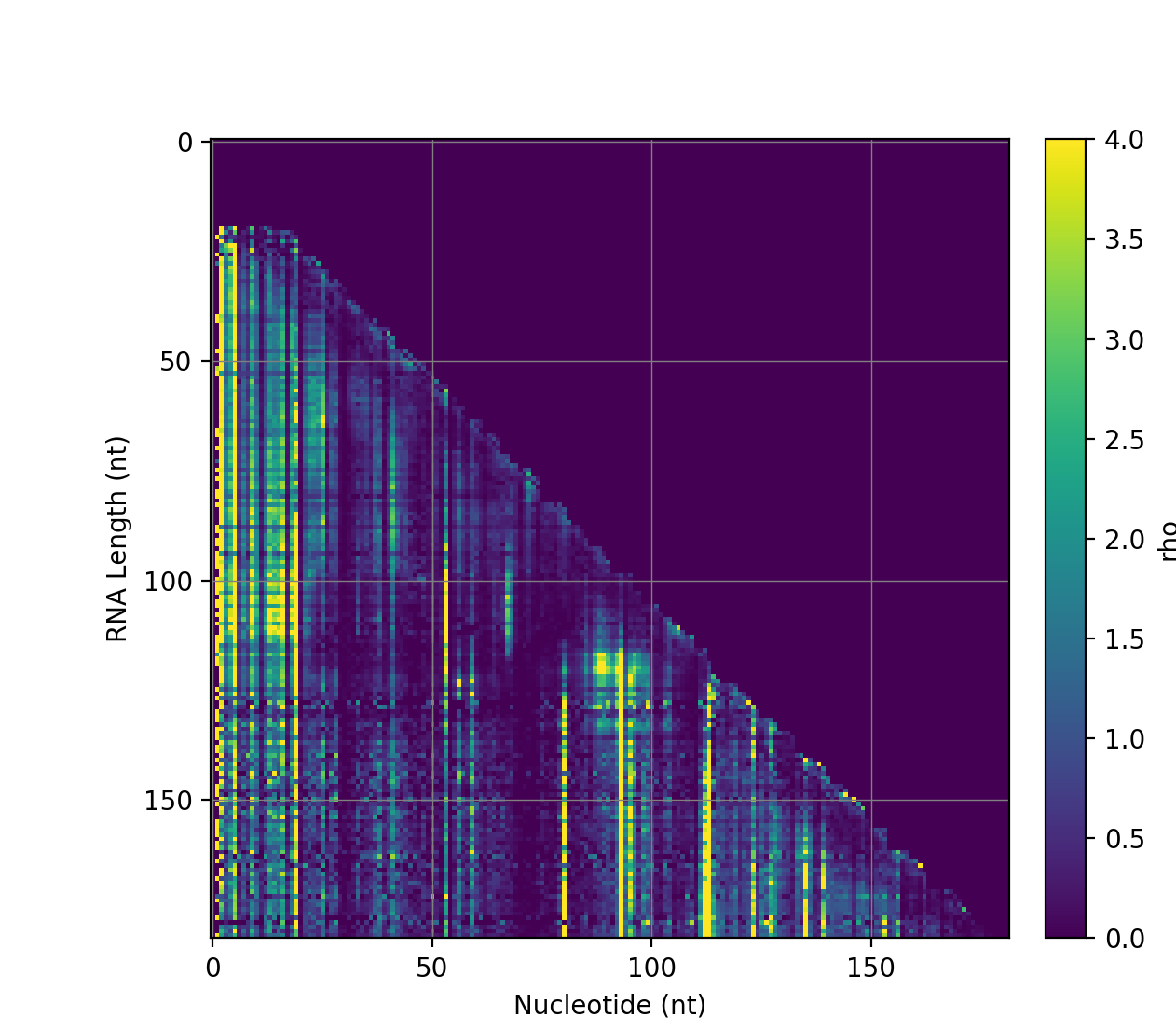
SPATS now includes automatic generation of reactivity plots,
visualization of sequence of alignments, Jupyter notebook integration,
and more.