After working with many different experimental setups across different labs, we've realized that there's a general problem in creating algorithms that properly collate and analyze experimental sequence data.
Our experience is that each experiment typically has its own custom software, involving things like trimming adapters, removing handles, and using a standard alignment tool (e.g., Bowtie) to determine if a particluar sequence matches the expected template.
The primary issue with systems like this is that they throw away information: knowing the experimental template gives you hints as to expected alignments, and can help correct for errors. We took this approach in our development of the SPATS tool, and our new sequence aligner now is capable of providing this for any type of experiment.
Let's take an example to illustrate. Typically, an experimental template looks something like this:

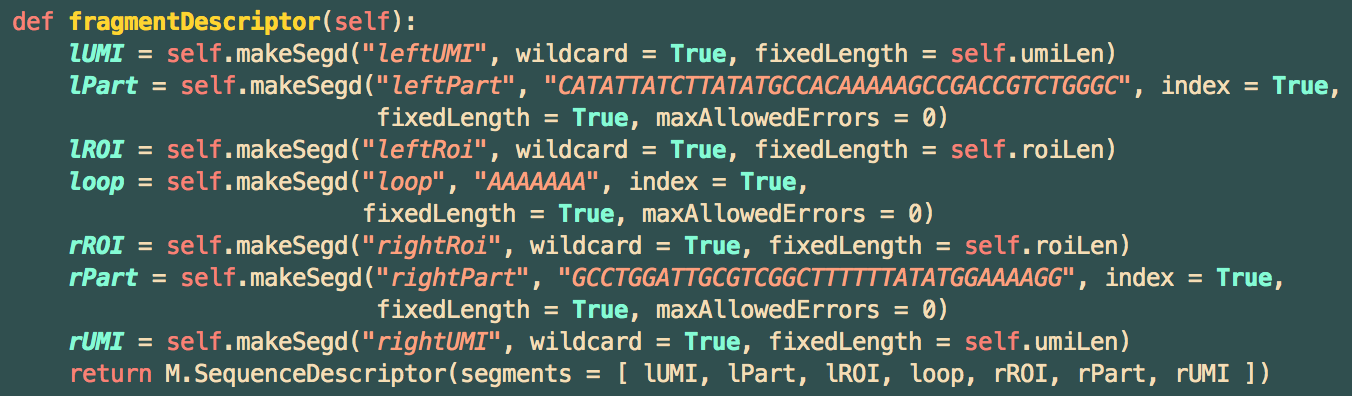
On the left (5') end is a 5-nt UMI, which can be any sequence of 5 nucleotides. This is followed by a 40-nt fixed sequence, and then another variable sequence, this time 3-nt in length. This is followed by a fixed loop of 7 A's, and then another 3-nt variable sequence, a length-36 fixed sequence, and finally another 5-nt UMI sequence.
This gets translated in code to an "experiment descriptor" as follows:
Our sequence-alignment code then uses this descriptor to analyze each R1/R2 pair, aligning based on the rules of each described segment. The sequence aligner can configurably handle mutations or indels in any of the segments. Here's an example of how the tool aligns to a target sequence:
As you can see, it first combines the R1/R2 pair into a single fragment, based on shared overlap (if present). Then, it has matched each of the described segments to a position on the fragment.
This is a relatively simple experiment, for ease of demonstration. Our engine also handles more complex experiments, including adapters that must be right- or left-aligned, multiple optional targets, and variable-length wildcard segments. Our highly optimized algorithms process millions of R1/R2 pairs per minute.
Once aligned, it's a simple matter to pull the desired metadata from an alignment (e.g., site/length of segment match, content of variable-nt segment) and collect counts for statistical processing.
Our code also automates barcode/UMI analysis, providing feedback on whether there was any bias in the amplification process. See our example barcode analysis.
Have a new experiment that needs alignment? Contact us for a free consultation.